
第一作者:浙江師範大學博士生劉曉豔,澳门京葡娱乐网博士生張銘元
通訊作者:楊啟華 教授,劉健 教授,高瑞 教授
通訊單位:浙江師範大學,澳门京葡娱乐网
論文DOI:https://doi.org/10.1021/acscatal.4c05501
全文速覽
氫的解離和遷移是加氫反應中的關鍵過程之一,但當反應底物含有硫原子時,這一過程變得極具挑戰,甚至不能發生,因為硫原子會嚴重毒化負載型金屬催化劑的活性金屬表面。本工作中,我們報道了一種在含硫狀态下仍能實現高效氫解離、氫轉移與選擇性加氫的催化策略,該方法通過氫傳遞物種與氫氣解離位點的協同作用,在Pt/MoO3催化含硫底物5-硝基苯并噻唑加氫反應中得以實現。Pt/MoO3在溫和反應條件下表現出99%的轉化率和~99%的選擇性,是迄今報道的含硫化合物加氫性能最高的催化劑之一。原位-XRD和DFT計算結果表明,Pt/MoO3催化劑的MoO3載體在反應中會儲存解離氫,形成的H1.68MoO3中間體具有獨特的H溢流和釋放性能,不僅加速了5-硝基苯并噻唑中N-O鍵的斷裂以實現加氫,同時還能有效覆蓋Pt NPs以防止其被硫毒化。該研究為解決當前含硫化合物催化加氫中的硫毒化挑戰提供了一種有前景的策略。
本文亮點:
1. 本工作設計制備的Pt/MoO3催化劑在含硫底物5-硝基苯并噻唑中表現出極高的催化活性,實現了99%的轉化率和>99%的選擇性。該結果在目前已報道的含硫化合物加氫催化劑中居于最活躍催化劑之列。
2. Pt/MoO3催化劑的H2解離位點和5-硝基苯并噻唑的硝基加氫活性位點分别位于Pt NPs和原位生成的H1.68MoO3,協同促進加氫的同時抗硫中毒。因此,Pt/MoO3具有1440 h⁻¹的高TOF,約為文獻報道值的三倍。
3. 機理研究表明活性物種H1.68MoO3在熱力學條件下不但易于形成,而且能夠将其存儲的氫自發釋放以還原−NO2基團。反應過程中,H1.68MoO3表面的快速氫轉移和固有氧空位是提升其加氫性能的關鍵因素。
圖文解析:
通過共沉澱-空氣煅燒法制備的xPt/MoO3催化劑,具有PtOx NPs分布在片狀結構的MoO3載體表面(圖1a-c),Pt含量分别為0.5,1,2和4 wt%。将其應用在含硫化合物5-硝基苯并噻唑加氫反應中,當反應溫度為80 ℃,氫氣壓力4 MPa,S/C=100(反應底物于Pt摩爾比)時(圖d-e),所有金屬含量的Pt/MoO3催化劑均具有優異的催化性能,其中2Pt/MoO3催化性能最優異,反應15 min轉化率>99%, TOF 為1440 h-1。值得注意的是,當反應條件更溫和時(40 ℃,2 MPa H2,S/C=200),2Pt/MoO3依然顯示了優異的催化性能,不過其反應初期出現了明顯的誘導期(圖1f),表明催化劑在反應期間發生了結構演化且生成了活性位點。
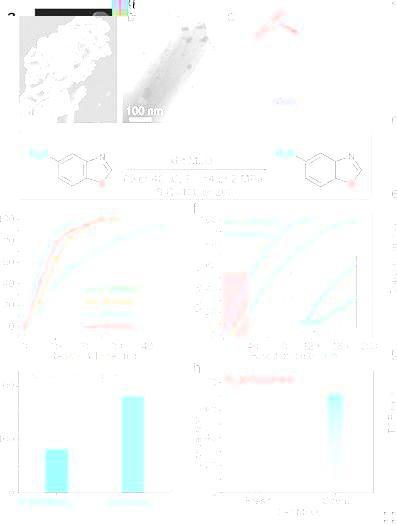
Pt/MoOx催化劑結構和催化性能
對反應後的最優催化劑2Pt/MoO3(命名為spent 2Pt/MoO3)進行表征後發現Pt NPs表面被覆蓋(圖2a-b),HRTEM,HAADF-STEM,EPR和XPS結果顯示該物質為HxMoO3物種(圖2c-f)。除此之外,将spent 2Pt/MoO3在氮氣氣氛下催化5-硝基苯并噻唑進行催化反應12 h(40 ℃,2 MPa N2, S/C=10)後成功得到了71.2%轉化率,而新鮮2Pt/MoO3催化劑卻不能在氮氣氣氛下發生催化反應(圖1h)。該結果進一步證明在反應過程中原位生成的HxMoO3為活性物種且可以作為H源。

反應後2Pt/MoO3結構表征
原位XRD表征結果顯示,Pt/MoO3在氫氣氣氛下會解離氫并通過氫溢流将活性H物種儲存到MoO3體相中形成H1.68MoO3(圖3),再次證明原位形成的H1.68MoO3是催化反應發生的活性物種。

催化劑結構演化表征
為了更深入地解析實驗結果,本研究采用密度泛函理論(DFT)對多種HxMoO3物相的穩定性,及其表面反應機理進行了詳細研究。首先,通過計算确定了HxMoO3晶體的最穩定結構(圖4a),進而計算了其在實驗反應溫度下的形成吉布斯自由能,結果發現H0.93MoO3為最穩定的物相,與實驗數據一緻。進一步的計算發現,HxMoO3晶體中的H原子結合能與其相轉變之間存在直接關聯(圖4b),特别是H0.93MoO3與H1.68MoO3之間的可逆性轉變。H原子在H1.68MoO3(010)表面上的遷移能壘計算(圖4c),進一步證明其快速的氫溢流使得H1.68MoO3成為了一種高效的氫儲存庫,有助于氫的釋放以促進加氫反應。吸附研究進一步顯示(圖4d),H1.68MoO3(010)表面更傾向于吸附5-硝基苯并噻唑的-NO₂端,有效避免了Pt表面的硫中毒。反應路徑分析揭示(圖4e),5-硝基苯并噻唑在H1.68MoO3(010)表面的加氫過程涉及N-O鍵解離形成表面*O和氫遷移脫除*O,并确定了通過H擴散在氧空位上脫除第二個*O原子形成*OH的過程為速率決定步驟,其勢壘為0.94 eV。綜上所述,由于H1.68MoO3(010)獨特的H質子儲存和釋放性能,不僅促進了5-硝基苯并噻唑氫化過程中N-O鍵的斷裂,而且有效防止了Pt的硫中毒,為開發更高效的催化劑和優化有機化合物氫化反應路徑提供了新的見解。
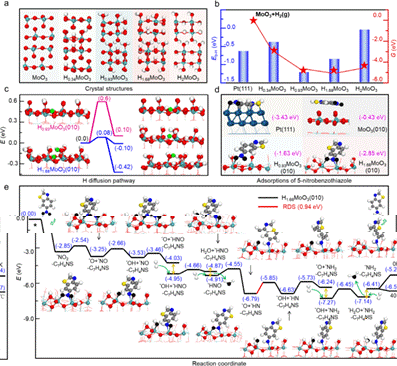
基于DFT研究Pt/MoO3原位演化過程和催化機理
總結與展望:
本工作展示了xPt/MoO3作為一種高效催化劑,可用于含硫化合物5-硝基苯并噻唑(5-nitrobenzothiazole)加氫為5-氨基苯并噻唑(5-aminobenzothiazole)的反應中,該産物是精細化學品和醫藥的重要中間體,xPt/MoO3表現出優異的抗S毒化性能。我們的實驗結果表明,H2和5-硝基苯并噻唑的活化位點分别為Pt和原位形成的H1.68MoO3,這一覆蓋在Pt表面的H1.68MoO3物種有效阻止了5-硝基苯并噻唑在Pt上的直接吸附,從而抑制了Pt的硫中毒。DFT計算進一步揭示,H1.68MoO3上自發的氫溢流和可逆相變對于硫化合物加氫至關重要,有助于防止Pt的硫中毒,提升5-硝基苯并噻唑的加氫活性,并作為氫存儲和傳遞的載體。綜上所述,實驗結果和理論模拟表明H2解離、氫溢流和H1.68MoO3的生成速率是影響xPt/MoO3催化性能的關鍵因素,這種通過包覆一層薄的H存儲和傳遞層,将H2解離和−NO2加氫活性位點分離開的辦法,可能是一種提升金屬催化劑在含O、N和S的不飽和化合物加氫反應中活性的有效策略。
